
|
||||||||||||||
| Autoren | ∞ | Werke | ∞ | Neu | ∞ | Information | ∞ | Shop | ∞ | Lesetips | ∞ | Textquelle | ∞ | |
|
|
||||||||||||||
| Autoren | ∞ | Werke | ∞ | Neu | ∞ | Information | ∞ | Shop | ∞ | Lesetips | ∞ | Textquelle | ∞ | |
Anzeige. Gutenberg Edition 16. 2. vermehrte und verbesserte Auflage. Alle Werke aus dem Projekt Gutenberg-DE. Mit zusätzlichen E-Books. Eine einmalige Bibliothek. +++ Information und Bestellung in unserem Shop +++
Wir beginnen unsere Betrachtungen mit einem einfachen Versuch. In ein Glas reinen Wassers schütten wir einen Löffel Kochsalz. Vom Kochsalz wissen wir, daß es eine Verbindung von Chlor und Natrium ist. Jedes Kochsalzmolekül besteht aus einem Natrium- und einem Chloratom, die chemisch fest miteinander verbunden sind.
Wir beobachten nun das Salz im Wasser und bemerken, daß das weiße Häufchen immer kleiner wird. Salzkorn auf Salzkorn verschwindet scheinbar, und wir sagen: Das Salz hat sich im Wasser gelöst. An Stelle des reinen Wassers haben wir jetzt eine Salzlösung. Durch verschiedene Proben können wir feststellen, daß das Salz sich tatsächlich vollkommen gleichmäßig in dem Wasser verteilt hat. Jedes Tröpfchen der Flüssigkeit zeigt einen salzigen Geschmack. Eine winzige Flüssigkeitsmenge der Lösung, an einer beliebigen Stelle entnommen und auf einer Nadelspitze in die Flamme des Bunsenbrenners gebracht, erregt dort die bekannte gelbe Natriumfärbung und liefert dadurch den Beweis, daß wirklich Chlornatriummoleküle in der Flüssigkeit vorhanden waren.
Wir müssen daher annehmen, daß die in das Wasser gebrachte Kochsalzmenge tatsächlich eine molekulare Verteilung erfahren hat, daß die Kochsalzmoleküle sich in dem Wasser ähnlich ausgebreitet haben, wie sich etwa die Moleküle eines Gases nach allen Seiten hin ausbreiten, sobald man es aus den einengenden Wänden seines Behälters entläßt. Mit gutem Grunde hat man deshalb auch diese Lösungsvorgänge mit den Ausbreitungsvorgängen eines sich selbst überlassenen Gases verglichen. Wie man dort von einem Gasdruck spricht, so redet man hier von einem Lösungsdruck. Dieser Vergleich hat sich als sehr fruchtbringend erwiesen, konnte doch der Chemiker van't Hoff nachweisen, daß dieser Druck genau so wie der Druck eines Gases den Gesetzen von Boyle und Gay-Lussac gehorcht. Van't Hoff konnte weiter zeigen, daß dieser osmotische oder Lösungsdruck auch der Größe nach vollkommen mit dem Gasdruck übereinstimmt.
Die Vorbedingung für alle diese Versuche war eine Membrane, welche zwar den Molekülen des Lösungsmittels, in unserem Falle also den Wassermolekülen, den freien Durchgang gestattete, ihn dagegen den Molekülen des gelösten Stoffes versperrte. Van't Hoff schaffte sich solche halb durchlässige Wände, indem er hart gebrannte poröse Tonzellen erst in eine Lösung von gelbem Blutlaugensalz und danach in eine Kupfervitriollösung eintauchte. Dabei bildete sich auf dem Ton eine feine Schicht von Ferrozyanidkupfer, welche die gewünschten Eigenschaften besaß. Eine derartig präparierte Tonzelle wurde nun mit irgendeiner Lösung gefüllt und mit einem undurchlässigen Pfropfen verschlossen, durch den ein langes Glasrohr in die Lösung hineinragte. Setzte man dann diese Zelle in ein Gefäß mit dem Lösungsmittel, in unserem Falle also in Wasser, so begann der osmotische Druck seine Wirkung zu äußern. Die Salzmoleküle wollten mit diesem Druck nach außen wandern, wurden aber durch die Kupferschicht daran gehindert. Mit dem gleichen Druck strebten die umgebenden Wassermoleküle in die Zelle hinein, und diese ließ die Kupferschicht frei passieren.
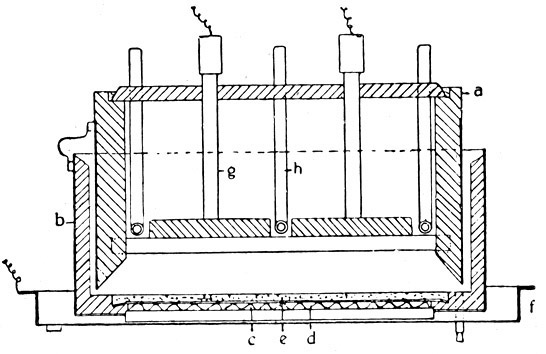
Fig. 100.
Schnitt durch einen Billiter-Apparat zur Elektrolyse von Kochsalzlösung in Ätznatron und Chlor.
Das ursprüngliche Billiter-Verfahren ist ein Glockenverfahren. Die Glocke (a), die den Anodenraum bildet, steht in einem Beton- oder Steinzeugkasten (b), dessen Boden von einem Eisendrahtnetz (c) gebildet wird. Auf dem Boden liegt ein Asbesttuch (d), das wieder als Träger für das Diaphragma (e) dient. Das Eisendrahtnetz steht einige Zentimeter über dem Boden eines Kastens (f), der an den negativen Pol der Stromquelle angeschlossen wird, während der positive Pol mit den Anoden (g), die im Deckel befestigt sind, verbunden wird; h ist ein Heizrohr, das horizontal zwischen je zwei Elektroden liegt.
Es kam also nichts aus der Zelle heraus, aber Wasser hinein, und die Flüssigkeit mußte deshalb in dem Glasrohr in die Höhe steigen. Aus der Höhe der Flüssigkeitssäule konnte man unmittelbar den osmotischen Druck ablesen. Mit dieser Apparatur vermochte van't Hoff nun die osmotischen Drucke der mannigfachsten Lösungen festzustellen. Das Ergebnis seiner Untersuchungen faßte er in einem Gesetz zusammen, welches sich dem Avogadroschen Gesetz an die Seite stellt.
Das Avogadrosche Gesetz besagt ja, daß in gleich großen Volumen verschiedener Gase, sofern sie nur bei gleicher Temperatur den gleichen Druck zeigen, stets die gleiche Anzahl von Gasmolekülen vorhanden ist. Das entsprechende Gesetz für die Lösungen besagt, daß in gleich großen Volumen der Lösungen verschiedener Stoffe stets die gleiche Anzahl von Molekülen dieser Stoffe vorhanden ist, falls die Lösungen bei gleichen Temperaturen die gleichen osmotischen Drucke zeigen. Durch dies van't Hoffsche Gesetz hatte man ein neues wertvolles Mittel zur Bestimmung der Molekulargewichte bekommen. Aber im weiteren Verlauf der Dinge zeigten sich bedauerliche Unstimmigkeiten. Namentlich bei schwachen Lösungen, bei denen also nur geringe Mengen des betreffenden Stoffes im Lösungsmittel vorhanden waren, zeigten sich osmotische Drucke von einer zunächst unerklärlichen Größe. Hatte das van't Hoffsche Gesetz seine Richtigkeit, dann mußten sich hier im Volumen des Lösungsmittels bedeutend mehr gelöste Moleküle vorfinden, als man nachweislich hineingetan hatte. Die Erklärung dieser Vorgänge gab der Chemiker Arrhenius in seiner Dissoziationstheorie. Damit aber wurde der Lösungsvorgang aus einem physikalischen zu einem chemischen Prozeß.
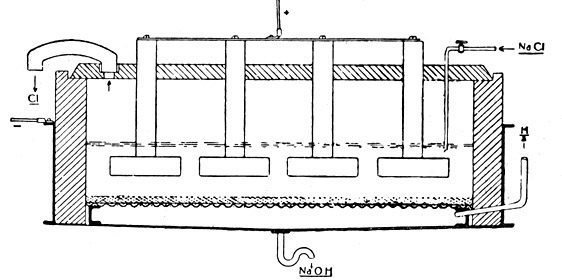
Fig. 101.
Das moderne Siemens-Billiter-Verfahren zur Zerlegung von Chlornatriumlösung in Chlor und Ätznatron.
Das Siemens-Billiter-Bad ist ein flacher Kasten aus Beton oder Steinzeug, der von einem Beton- oder Steinzeugdeckel, in dem die Anoden befestigt sind, abgeschlossen wird. Im Deckel befinden sich noch ein Steinzeugkrümmer (links), der zum Abzug des Chlorgases dient, ein Glas- oder Tonrohr, durch das die frische Salzlösung zugeführt wird (Mitte, unten). Einige Zentimeter über dem Boden ist ein Eisendrahtnetz wagerecht gelagert, das als Träger für ein Asbesttuch und gleichzeitig als Kathode dient. Auf das Asbesttuch wird als Diaphragma eine Paste von Bariumsulfat und Asbest in dünner Schicht aufgeschlämmt. Dicht unterhalb des Eisendrahtnetzes liegen wagerecht Eisenrohre, die den Abzug des freien Wasserstoffes gestatten.
Bis dahin hatte man die Lösung im Gegensatz zur chemischen Verbindung durchaus als einen rein physikalischen Vorgang angesehen. Konnte man doch durch einfache physikalische Mittel den gelösten Stoff vollkommen unverändert zurückerhalten. Brachte man beispielsweise die eingangs erwähnte Kochsalzlösung über ein Feuer, so verdampfte das Wasser, und man erhielt das in die Lösung gegebene Kochsalz als Rückstand. Es war eben eine molekulare Auflösung, aber das einfache Molekül blieb dabei unverändert. Nach der Erklärung von Arrhenius wurde dies anders. Nach seiner Auffassung fand namentlich bei schwachen Lösungen auch eine teilweise Dissoziation oder Spaltung der Moleküle des gelösten Stoffes statt. Bei der Kochsalzlösung mußten sich also einzelne Salzmoleküle in Natriumteilchen und Chlorteilchen trennen, die unabhängig voneinander im Lösungsmittel umherschwammen.

Fig. 102.
Chlor-Ätznatron-Anlage für eine Tagesproduktion von etwa 1000 kg Chlorkalk, ausgeführt für die Sulfitzellulosefabrik Pötschmühle der Firma Ignaz Spiro & Söhne, Krummau a. d. Moldau.
Die einzelnen Elektrolyseure der Batterien sind nach dem in Fig. 101 gegebenen Schema gebaut.
Diese neue Lehre erfuhr bei ihrem ersten Auftauchen gerade von seiten der älteren Chemiker den schärfsten Widerspruch. In der Tat schien sie auch allen bisherigen Erfahrungen ins Gesicht zu schlagen. Bringt man Natrium in Wasser, so gibt es ja sofort eine Wasserzersetzung, Wasserstoff entweicht, und das Natrium bildet unter Aufnahme eines Oxylradikals OH Ätznatron von der Form NaOH, welches sich im Wasser löst. Leitet man aber Chlorgas in Wasser, so wird es vom Wasser in erheblichen Mengen absorbiert und bildet das bekannte Chlorwasser mit starkem Chlorgeruch. Nichts indes von diesen beiden Dingen war bei einer einfachen Kochsalzlösung zu beobachten, in der ja doch nach Arrhenius freie Chlor- und Natriumteilchen vorhanden sein sollten.
Aber die Dissoziationstheorie nimmt ja auch nicht an, daß die Moleküle des gelösten Stoffes in ihre Atome, sondern daß sie in Ionen gespalten werden. Das Ion unterscheidet sich vom einfachen Atom oder auch von einer einfachen Atomgruppe durch eine positive oder negative elektrische Ladung. Nach der Art der Ladung gibt es positive und negative Ionen. In unserem Falle des Kochsalzes sind Kochsalzmoleküle in positive Natriumionen und in negative Chlorionen getrennt. Mit dieser Annahme führt die Dissoziationstheorie aber zu jenen elektrochemischen Anschauungen, die bereits um die Mitte des vorigen Jahrhunderts von Berzelius entwickelt wurden. Nach dieser Anschauung gibt es ausgesprochen positive und negative Elemente, deren stärkere oder schwächere chemische Verwandtschaft auf elektrischen Anziehungen ihrer Atome infolge ungleichnamiger Ladungen beruht. Die Ionen der Elemente unterscheiden sich nun nach der genannten Theorie sehr stark von einfachen neutralen Atomen. Das Natriumion beispielsweise mit einer positiven elektrischen Elementarladung kann ruhig im Wasser schwimmen, ohne irgendwelche chemischen Reaktionen auszuüben. Das gleiche gilt vom Chlorion, welches ein überschüssiges Elektron, also eine negative Elementarladung, besitzt.
Das Bild ändert sich aber sehr stark, wenn wir zwei Platinbleche in unsere Salzlösung stecken und mit einer Stromquelle verbinden. Folgen wir der Richtung des negativen Stroms, wandern wir also in der Richtung des Elektronenflusses, als welchen wir ja jeden elektrischen Strom auffassen. Dann wird sich zunächst das mit dem negativen Pol unserer Stromquelle verbundene Platinblech mit Elektronen füllen, also negative elektrische Ladung zeigen. Infolgedessen wird es alle in seiner Nähe befindlichen positiven Natriumionen anziehen, wie sich ungleichnamige Elektrizitätsmengen nach dem Coulombschen Gesetz anziehen müssen. Sobald die Natriumionen aber das negativ geladene Platinblech berühren, gleichen sich die Ladungen aus. Aus dem Blech springt ein Elektron zu dem Natriumion hin, und im gleichen Moment verwandelt sich dies in ein neutrales Natriumatom mit allen Eigenschaften eines solchen. Unter anderen Verhältnissen würde es sich mit einem zweiten Natriumatom zu einem Natriummolekül vereinigen. Da es sich hier aber im Wasser befindet, so kommt es gar nicht zu dieser Molekülbildung, sondern es treten sofort die uns vom Natrium bekannten chemischen Reaktionen ein. Es bilden sich unter Aufspaltung eines Wassermoleküls H2O freier Wasserstoff und Ätznatron NaOH, welch letzteres sofort in Lösung geht.
Der entsprechende Vorgang spielt sich an dem anderen, mit dem positiven Pol der Stromquelle verbundenen Platinblech ab. In diesem Blech herrscht Elektronenmangel, da die Stromquelle die Elektronen von hier zu sich hinsaugt. Das Blech zeigt also positive Ladung und zieht die negativen Chlorionen zu sich heran. Bei der Berührung mit dem Blech geben diese ihre überschüssigen Elektronen an das Blech ab und sind im gleichen Moment zu neutralen Cloratomen geworden, die sich zu Chlormolekülen vereinigen und im Wasser gelöst bleiben. Sobald wir also auf unsere Lösung einen Strom wirken lassen und eine Elektrolyse vornehmen, treten tatsächlich alle diejenigen Erscheinungen ein, die man bereits bei einer einfachen Lösung erwarten müßte, wenn eben nicht bei dieser eine Spaltung der Moleküle in Ionen, sondern in Atome stattfände.

Fig. 103.
Elektrolyseur der Siemens & Halske A.-G. zur Zerlegung von Kochsalzlösung zwecks Gewinnung von Natriumhypochlorit.
Die Elektrolyse vollzieht sich nach den Formeln:
1.
| Elektrolyt 2 NaCl Kochsalz (Chlornatrium) |
= |
Kathode 2 Na Natrium |
+ |
Anode Cl2 freies Chlor |
2.
| 2 Na Natrium |
+ | 2 H2O Wasser |
= | 2 NaOH Natriumhydroxyd (Ätznatron) |
+ | H2 gasförmig entweichen- der Wasserstoff |
3.
| 2 NaOH Natriumhydroxyd (Ätznatron) |
+ | Cl2 Chlor |
= | NaCl Kochsalz |
+ | H2O Wasser |
+ | NaOCl unterchlorigsaures Natrium (Natrium- hypochlorit) |
Das Natriumhypochlorit dient zur Reinigung von Wasser, besonders in den großstädtischen Badeanstalten. (Chlorreinigung.)
Die Frage ist zu beantworten, wie sich die Dinge nun weiter entwickeln, wenn wir den Strom längere Zeit auf unsere Lösung wirken lassen. Nach der kinetischen Wärmetheorie herrscht in unserer Lösung, die ja eine gewisse Temperatur besitzt, eine fortwährende, recht beträchtliche, unregelmäßige Bewegung der kleinsten Teilchen. Positive und negative Ionen stoßen zusammen und vereinigen sich wieder zu neutralen Molekülen. Andere Moleküle prallen aufeinander und spalten sich in Ionen. In diese zunächst vollkommen regellose und nach keiner Seite gerichtete Bewegung bringt nun aber der elektrische Strom eine gewisse Ordnung und Richtung. Alle positiven Ionen werden von dem positiven Platinblech abgestoßen, von dem negativen angezogen. Umgekehrt ergeht es den negativen Ionen.
Es ist also eine Wanderung dieser Ionen in der Lösung zu erwarten. In früheren Jahrzehnten, vor dem Aufkommen der Dissoziationstheorie, glaubte man nicht an eine solche Bewegung. Man nahm an, daß das negative Blech dem nächstgelegenen Salzmolekül ein positives Natriumion entrisse und neutralisierte, während das übrigbleibende negative Chlorion nun seinerseits wieder dem nächsten Salzmolekül das Natrium entzöge. Nach dieser Anschauung bliebe also jedes Molekül an seiner Stelle, und nur dadurch, daß jedesmal die rechte Hälfte des nten Moleküls mit der linken Hälfte des n + 1ten Moleküls ein neues Molekül bilde, würden die entsprechenden Molekülhälften, also in unserem Falle Natrium und Chlor, an den beiden Elektroden fortwährend und in beträchtlichen Mengen frei. Es ist wahrscheinlich, daß auch dieser Vorgang bei der Elektrolyse stattfindet. Daß aber außerdem auch die bereits erwähnte Ionenwanderung in jedem Elektrolyt stattfindet, ist sicher erwiesen, und auch die Geschwindigkeit dieser Wanderungen ist festgestellt. Nachstehend seien einige Geschwindigkeiten nach den Angaben von Dr. Ries gegeben.
| a) Positive Ionen | ||
| H (Wasserstoffionen) | 0,00300 | cm pro Sekunde |
| K (Kaliumionen) | 0,00057 | " |
| Na (Natriumionen) | 0,00035 | " |
| NH4 (Ammoniumionen) | 0,00055 | " |
| Ag (Silberionen) | 0,00046 | " |
| b) Negative Ionen |
||
| OH (Hydroxylionen) | 0,00156 | cm pro Sekunde |
| Cl (Chlorionen) | 0,00059 | " |
| J (Jodionen) | 0,00061 | " |
| NO3 (Salpetersäurerestionen) | 0,00053 | " |
| C2H3O2 (Essigsäurerestionen) | 0,00029 | " |
Die vorstehende Tabelle zeigt uns nicht nur die Ionen einzelner Elemente, wie der salzbildenden Stoffe Chlor und Jod sowie der Alkalimetalle Kalium und Natrium, sondern auch ionisierte Atomgruppen, wie beispielsweise das Hydroxyl OH. Solche eng verknüpften Atomgruppen lernten wir bereits bei der Betrachtung der molekularen Struktur verschiedentlich kennen und bezeichneten sie als Radikale. Aus der vorstehenden Tabelle sehen wir, daß das Oxylradikal stets als elektronegatives Ion auftritt, während der Wasserstoff immer als positives Ion in die wäßrige Lösung geht.
Durch diese Beobachtung und Untersuchung der Vorgänge bei der Lösung im Wasser mit darauffolgender Elektrolyse ist man heute auch in der Lage, gewisse chemische Typen besser zu definieren und in ihren Wirkungen zu erklären, als es vordem der Fall war. Die Chemie unterscheidet bekanntlich zwei große Gruppen von Verbindungen, die Basen und die Säuren. Rein äußerlich erkennt man diese Stoffe durch ihre Einwirkung auf Lackmuspapier, wobei die einfache Gedächtnisregel gilt: Base bläut, Säure rötet. Der Zusammensetzung nach betrachtet man die Base als Verbindung eines Metalls mit einer Oxylgruppe, gibt ihr also die allgemeine Form MOH, wobei M ein Metall bedeutet. Im Gegensatz dazu wird die Säure als die Verbindung einer Atomgruppe mit einem Wasserstoffatom angesehen von der allgemeinen Form RH wobei R den Säurerest bedeutet. Bei jeder Lösung in Wasser müssen bei einer Base negative Hydroxylionen, bei einer Säure positive Wasserstoffionen auftreten, und man führt heute alle die charakteristischen Wirkungen der beiden Verbindungstypen auf die Tätigkeit der HO- oder H-Ionen zurück.
Schon die Alchimisten des Mittelalters hatten den Satz aufgestellt: Corpora non agunt nisi fluida, d. h. auf deutsch: Die Körper reagieren nur in flüssigem Zustande, sei es gelöst oder geschmolzen. Die Wahrheit dieses Wortes ist uns jetzt klar, nachdem wir die Wirkung der Lösung im Wasser, den Zerfall gelöster Moleküle in positive und negative Atomgruppen oder Atome, eben in jene Ionen, kennengelernt haben. Bringen wir etwa eine basische Verbindung MOH und eine Säure RH in demselben Wasser in Lösung, so tritt sofort die teilweise Dissoziation ein. Die Säure zerfällt in ein positives Wasserstoffion und den negativen Säurerest R, die Base in ein negatives Oxylion OH und ein positives Radikal oder Metall M. Wasserstoffion H und Hydroxylion OH vereinigen sich sofort zu Wasser H2O. Das negative Radikal der Säure R und das positive Radikal der Base M schließen sich dagegen zu einem Salz von der Form MR zusammen.
Man unterscheidet nun in der Chemie bekanntlich stärkere und schwächere Basen und Säuren. Eine exakte Definition dieses Begriffes vermochte man aber vor der Ergründung der Lösungserscheinungen nicht zu geben. Man bezeichnete diejenige Säure als die stärkere, welche die andere Säure aus ihren Verbindungen zu vertreiben vermochte, heute läßt sich dieser Begriff aber auch zahlenmäßig definieren. Die stärkere Säure bzw. Base ist immer diejenige, von der bei einer Lösung in Wasser unter sonst gleichen Verhältnissen ein größerer Prozentsatz ihrer Moleküle dissoziiert wird. Denn wir müssen ja immer daran festhalten, daß bei einer Lösung keineswegs die gesamte Anzahl der gelösten Moleküle in Ionen aufgespalten wird, sondern ein gewisser Prozentsatz, der einmal von der Art des gelösten Stoffes, weiter aber auch von der Temperatur und von der Stärke der Lösung abhängt. Im allgemeinen sind in einer sehr schwachen Lösung prozentual mehr Moleküle dissoziiert als in einer starken.
Normalerweise bezieht man alle Angaben über den Grad der Dissoziation auf Normallösungen von 18 Grad Celsius. Unter einer Normallösung versteht man dabei eine Lösung, welche in einem Liter Wasser ein Grammol des gelösten Stoffes enthält. Das Grammol bedeutet dabei so viel Gramm, wie das Molekulargewicht des betreffenden Stoffes beträgt. Man hat nun beispielsweise festgestellt, daß in einer Normallösung von Flußsäure FlH 3 Prozent aller Moleküle dissoziiert sind. Dagegen ist die prozentuale Dissoziation der Salzsäure wesentlich größer, und auch das Natriumsalz der Flußsäure besitzt einen höheren Dissoziationsgrad. Wir vermengen nun zwei Normallösungen von Fluornatrium und Salzsäure. Das Fluornatrium liefert in diesen positive Natriumionen und negative Fluorionen, die Salzsäure positive Wasserstoffionen und negative Chlorionen. Es befinden sich daher in der gemischten Lösung die beiden Komponenten der Flußsäure, Wasserstoff- und Fluorionen, in einem Prozentsatz, der über die angegebenen 3 Prozent beträchtlich hinausgeht. Daher muß eine Rückbildung dieser Ionen in Form ihrer Vereinigung zu Flußsäure von der Form HFl stattfinden, während Natrium- und Chlorionen, die Komponenten des Kochsalzes, welche zu höherem Prozentsatz als freie Ionen in der Lösung existieren können, übrigbleiben. Äußerlich stellt sich aber dieser Vorgang so dar, als ob die stärkere Salzsäure die schwächere Flußsäure aus ihrem Salz vertrieben hat.
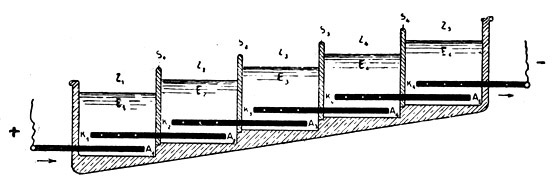
Fig. 104.
Batterie zur Gewinnung von Natriumhypochlorit aus Kochsalzlösung im großen.
Z Zellen, K Kathoden, A Anoden, E Elektrolyt, S Scheidewände.
Die einzelnen Zellen sind in der großen Zahl hintereinandergeschaltet, daß die ganze Einrichtung mit dem gewöhnlichen Netzstrom der Lichtleitung betrieben werden kann.
Wir wollen nun verschiedene Lösungen den Wirkungen der Elektrolyse aussetzen und die dabei auftretenden Erscheinungen untersuchen. Wir wählen zu diesem Zweck eine Lösung von Kupfervitriol SO4Cu in Wasser. Das Kupfervitriol, auch schwefelsaures Kupfer genannt, ist ein Salz. Es enthält in Übereinstimmung mit dem vorstehend gegebenen Schema das Radikal der Schwefelsäure H2SO4 von der Form SO4 und das metallische Radikal einer Base, nämlich Kupfer Cu. Bei der Lösung muß sich ein Teil der Vitriolmoleküle in negative Ionen von der Form SO4 und in positive Kupferionen von der Form Cu spalten. Die Atomgewichte für die Bestandteile sind 32 für Schwefel, 16 für Sauerstoff und 63,5 für Kupfer. Einem Kupferion vom Gewicht 63,5 entspricht daher ein Säureion SO4 vom Gewicht 32 + 4 x 16 = 96. Bei der Elektrolyse wird das positive Kupfer an die Kathode wandern, der negative Rest SO4 an die positive Anode, und zwar treten diese beiden Verbindungen genau in den Gewichtsverhältnissen 63,5 zu 96 auf. Das Kupfer schlägt sich als metallisches Kupfer an der Kathode nieder. Während das Natrium bei unserem früheren Versuche sich sofort im Wasser zersetzte, kann ja das Kupfer im Wasser ohne weiteres bestehen und bildet den bekannten galvanischen Niederschlag. Etwas anderes ist es dagegen mit dem Schwefelsäureradikal SO4. Dies kann wohl als negatives Ion in der Lösung existieren, aber nicht mehr als neutralisiertes Molekül. Es braucht dringenden Ersatz für das zweiwertige, ihm bei der Elektrolyse abhanden gekommene Kupfer und sucht ihn, wo es ihn findet. Besteht die Anode aus Kupferblech, so ist die Angelegenheit verhältnismäßig einfach. Infolge des Lösungsdruckes, welchen Kupfer in Kupfervitriol besitzt, treten positive Kupferionen in die Lösung und vereinigen sich mit dem Säurerest SO4 wieder zu Kupfervitriolsalz. Sofern überhaupt ein Anodenmetall vorhanden ist, dessen Lösungsdruck den widerstehenden osmotischen Druck der Flüssigkeit zu überwinden vermag, gehen Atome davon in die Lösung und bilden mit dem Säurerest SO4 ein Salz. Besteht die Anode beispielsweise aus Eisen, so bildet sich Eisenvitriol von der Form FeSO4. Aber das Bild ändert sich, wenn ein Metall von sehr geringem Lösungsdruck, beispielsweise Platin, für die Anode genommen wurde. Dann findet der Säurerest SO4 keine Metallionen.
Ist dies der Fall, dann wirkt sich eine andere Erscheinung, nämlich die Ionisierung des Wassers, selbst aus. Auch in reinem Wasser sind stets einige der H2O-Atome in positive Wasserstoffionen und negative Hydroxylionen gespalten. Die Zahl dieser Ionen ist sehr gering. Sie ist nach verschiedenen Methoden übereinstimmend dahin gefunden worden, daß in 12 000 Kubikmeter Wasser 1 Gramm H-Ionen und 17 Gramm Hydroxylionen vorhanden sind. Das ist gewiß nicht allzuviel. Aber erstens bilden sich automatisch sofort neue Ionen dieser Art, sowie einige der vorhandenen aufgebraucht und anderweitig gebunden werden. Andererseits sind die negativen Hydroxylionen bereits durch die elektrische Anziehung in der Nähe der positiven Anode in besonderer Dichte versammelt. Sie befinden sich aber nun in einer ganz ähnlichen Lage, wie der Säurerest SO4. Sobald sie nämlich durch Abgabe ihrer negativen Ladung an die Anode ihren Ionencharakter verlieren und einfache Oxylradikale werden, können sie nicht mehr frei existieren. Es könnten sich zur Not zwei Oxylradikale zu einem Wasserstoffsuperoxydmolekül von der Form H2O2 verbinden. Aber auch diese Verbindung wäre wenig beständig und würde sich sehr bald freiwillig in Sauerstoff und Wasser aufspalten. Tatsächlich tritt denn auch etwas anderes ein.
Das Oxylion besteht ja aus einem Wasserstoffatom mit einer elektrischen positiven Elementarladung und einem Sauerstoffatom mit zwei negativen elektrischen Ladungen. Durch ihre Vereinigung zum Hydroxyl wird die eine Ladung des Sauerstoffs von der entgegengesetzten Ladung des Wasserstoffs neutralisiert, eine negative Ladung bleibt hingegen wirksam übrig. In nächster Nähe der positiven Anode wirkt nun eine sehr starke anziehende Kraft auf den negativen Sauerstoff und eine starke abstoßende Kraft auf den positiven Wasserstoff. Der Effekt dieser verschieden gerichteten Kräfte auf die beiden Bestandteile des Hydroxyls äußert sich schließlich darin, daß es zerreißt.
Ein positives Wasserstoffion wird frei und von der Anode abgestoßen, während ein negatives Sauerstoffion ihr zueilt, seine Ladung abgibt und zum neutralen Sauerstoffatom wird.

Fig. 105.
Elektrodensatz für die Kupferraffinerie durch Elektrolyse.
Die Anoden bestehen aus dicken Platten gegossenen Schwarzkupfers, das sich auflöst und als chemisch reines Kupfer auf den Kathoden niederschlägt. Die Kathoden bestehen aus dünnen Blechen aus reinem Kupfer. Das Elektrolyt ist eine Kupfervitriollösung mit geringem Schwefelsäurezusatz.
Da es aber als solches nicht frei existieren kann, verbindet es sich sofort mit einem anderen Sauerstoffatom zum zweiatomigen Sauerstoffmolekül, es tritt also freier Sauerstoff O2 in Bläschenform an der Platinanode auf. Dagegen vereinigen sich die abgestoßenen positiven Wasserstoffionen mit dem negativen Säurerestion SO4 zu neutralen Schwefelsäuremolekülen von der Form H2SO4. Wir müssen uns noch erinnern, daß bei der teilweisen Dissoziation des Wassers jedem Hydroxylion ein Wasserstoffion entsprach, und daß diese positiven Wasserstoffionen sich in der Umgebung der negativen Kathode ansammeln müssen. Aber sie finden hier keine Gelegenheit zur chemischen Betätigung, geben ihre positive Ladung an die Kathode ab und treten als freier Wasserstoff von der Form H2 bläschenförmig an der Kathode auf.
Nach dem bisher Gesagten haben wir also folgende Erscheinungen: Setzen wir eine Kupfervitriollösung der Elektrolyse zwischen zwei Kupferelektroden aus, so löst sich einfach Kupfer an der Anode und schlägt sich in gleicher Menge an der Kathode nieder. Nehmen wir dagegen eine Kupferkathode und eine Eisenanode, so schlägt sich Kupfer auf der Kathode nieder, es verschwindet die dem niedergeschlagenen Kupfer äquivalente Kupfervitriolmenge aus der Lösung. Dafür geht aber von der eisernen Anode her eine äquivalente Eisenmenge in die Lösung und bildet eine entsprechende Menge Eisenvitriol. Nach einer gewissen Zeit wird sämtliches Kupfervitriol aus der Lösung verschwunden sein. Dann beginnt sich aus dem Eisenvitriol Eisen auf der Kathode niederzuschlagen, und wir haben jetzt praktisch zwei eiserne Elektroden in Eisenvitriollösung. Wird dagegen die Anode aus Platin gewählt, so schlägt sich zwar ebenso wie bei den anderen Versuchen Kupfer auf der Anode nieder. Dagegen geht gleichzeitig mit diesem Niederschlag eine Zersetzung des in der Lösung vorhandenen Wassers in Sauerstoff und Wasserstoff einher. Der stets positive Wasserstoff tritt bläschenförmig an der Kathode, der elektronegative Sauerstoff an der Anode auf. Dieser Vorgang vollzieht sich so lange, bis jedes Kupfervitriolmolekül der Lösung sein Kupfer an die Kathode abgegeben hat und in ein Schwefelsäuremolekül verwandelt worden ist. Danach stehen die Elektroden in verdünnter Schwefelsäure und die weiteren Erscheinungen beschränken sich auf eine reine Wasserzersetzung.
Betrachten wir nun die Verhältnisse zwischen den an den Elektroden ausgeschiedenen chemischen Stoffen und den für die Elektrolyse verbrauchten Elektrizitätsmengen. Wir sahen bereits, daß diese für jedes abgeschiedene Atom bzw. Molekül keineswegs gleich sind. Das Sauerstoffion brauchte die doppelte Menge positiver Elektrizität zu seiner Neutralisation wie das Wasserstoffion an negativer Elektrizität. Es mußte also das Sauerstoffatom zwei Elektronen an die Anode abgeben, um neutralisiert zu werden, das Wasserstoffion mußte zu dem gleichen Zweck ein Elektron aus der Kathode herausziehen.
Nun ist aber nach einem Grundgesetz der Elektrizitätslehre die Stromstärke in allen Querschnitten eines unverzweigten Stromkreises gleich groß, d. h. es laufen durch jeden Querschnitt einer elektrischen Strömung in der Zeiteinheit gleich viel Elektronen. Daraus folgt, daß sich bei einer reinen Wasserzersetzung an der Kathode die doppelte Anzahl von Wasserstoffionen zu Wasserstoffatomen neutralisieren und ausscheiden müssen, wie Sauerstoffionen an der Anode. Und da in beiden Fällen je zwei Atome sich zu einem Wasserstoff- bzw. Sauerstoffmolekül vereinigen, so müssen die doppelte Anzahl von Wasserstoffmolekülen an der Kathode auftreten, wie Sauerstoffmoleküle an der Anode. Und da schließlich das Volumen eines Gases bei gleicher Temperatur und gleichem Druck proportional der Anzahl seiner Moleküle ist, so muß das Volumen des an der Kathode frei werdenden Wasserstoffs das Doppelte des an der Anode entwickelten Sauerstoffs betragen, was mit den Beobachtungen durchaus übereinstimmt.

Fig. 106.
Blick in eine große Kupferraffinerie.
Ein mechanischer Kran besorgt die notwendige Auswechselung der Elektroden. Aufgezehrte Anoden werden durch neue ersetzt, die stark gewordenen Kathoden herausgehoben und zur Schmelze gebracht. Die Arbeiter neben der Elektrode am Kran geben einen Begriff von den Größenverhältnissen.
Wir sehen hier, daß dieselbe Strommenge nicht nur volumenmäßig, sondern auch dem Gewichte nach an den beiden Elektroden sehr verschiedene Mengen der einzelnen Elemente ausscheidet. Aber diese Mengen sind keineswegs regellos, sondern stehen in einem bestimmten Äquivalentsverhältnis. Zur genaueren Feststellung dieser elektrochemischen Äquivalente müssen wir uns zunächst über die Maßeinheit der Elektrizitätsmenge einigen. Wir gehen dabei von der praktischen Einheit der Stromstärke, dem Ampere aus. Ein elektrischer Strom besitzt die Einheit der Stromstärke oder fließt mit der Stärke von einem Ampere, wenn er in jeder Sekunde die Elektrizitätsmenge von einem Coulomb durch den Querschnitt des Stromlaufes sendet.
Man hat nun mit größter Genauigkeit elektronische Zersetzungsversuche mit reinen Silberelektroden in einer Lösung salpetersauren Silbers von der Form AgNO3 angestellt und dabei gefunden, daß ein Strom von einem Ampere in einer Sekunde 0,00118 Gramm Silber auf der Kathode niederschlägt. Nun beträgt das Atomgewicht des einwertigen Silbers 107,88. Ein Grammol oder ein Grammäquivalent Silber besitzt demnach ein Gewicht von 107,88 Gramm, und da man für die Niederschlagung von 0,00118 Gramm Silber ein Coulomb benötigt, so braucht man für das Grammäquivalent Silber 107,88 : 0,00118 = 96 494 Coulomb.
Diese Zahl ist in der Elektrochemie von Wichtigkeit, denn sie gilt auch für das Grammäquivalent eines jeden anderen Stoffes. Das Grammäquivalent aber wird erhalten, indem man das Atomgewicht durch die Wertigkeit des betreffenden Stoffes dividiert. Das Atomgewicht des Wasserstoffs ist gleich 1 und seine Wertigkeit ebenfalls. Hier beträgt also das Grammäquivalent tatsächlich 1 Gramm, und wir können nun sagen, daß 1 Gramm Wasserstoff in einem elektronischen Prozeß beim Durchgange von 96 494 Coulomb ausgeschieden wird. Das Atomgewicht des Sauerstoffs beträgt hingegen 16, und seine Wertigkeit ist 2. Das Grammäquivalent Sauerstoff umfaßt daher 8 Gramm Sauerstoff, und wenn wir die bei dem früher beschriebenen Versuch entwickelten Wasserstoff- und Sauerstoffmengen sorgfältig auffangen und wiegen, so werden wir in der Tat finden, daß dem Gewicht nach immer 8mal so viel Sauerstoff wie Wasserstoff entwickelt wird.
Nach der gegebenen Regel lassen sich nun ohne Schwierigkeiten die Äquivalenzgewichte aller anderen Stoffe und Radikale berechnen. Chlor hat beispielsweise das Atomgewicht 35,5 und ist einwertig. Hier beträgt das Grammäquivalent also 35,5 Gramm. Kupfer mit dem Atomgewicht 63,57 und zwei Valenzen hat das Äquivalentgewicht 31,78 Gramm, Aluminium mit dem Atomgewicht 27 und drei Valenzen das Äquivalentgewicht 9. In der Praxis wird man freilich umgekehrt vorgehen. Man wird das zu untersuchende Element unter Aufwendung einer genau gemessenen Anzahl von Coulombs niederschlagen, wird die niedergeschlagene Menge durch äußerst genaue Wägungen ermitteln und danach das Aquivalentgewicht feststellen. Da das Aquivalentgewicht aber nichts anderes als das Atomgewicht, dividiert durch die Wertigkeit, ist, und da die Wertigkeit immer eine ganze Zahl sein muß, so geben solche elektronischen Versuche ein wertvolles Hilfsmittel zur Bestimmung der Atomgewichte und der Wertigkeiten. Nach der Stellung des Kupfers im periodischen System der Elemente hätte man erwarten können, daß das Kupfer einwertig sein müsse. Die Elektrolyse beweist indes seine Zweiwertigkeit.
Wir gingen bei der Untersuchung der elektrolytischen Vorgänge zunächst von den Erscheinungen bei der Lösung irgendeines Stoffes in Wasser aus und lernten dabei die Dissoziation, die teilweise Zerlegung von Molekülen in Ionen kennen. Wir sahen, daß irgendein positives oder negatives Ion sich in chemischer Beziehung infolge des Fehlens oder Zuviels einer oder mehrerer elektrischer Elementarladungen sich ganz gewaltig von dem einfachen Atom unterscheidet. Die gleichen Erscheinungen treten nun aber auch beim Schmelzen der Substanzen auf. Es ist gleichgültig, ob man einen Stoff durch Lösung oder durch Erwärmung in den flüssigen Zustand zersetzt. In beiden Fällen findet eine teilweise Dissoziation statt, und sowohl Lösungen wie Schmelzen sind daher für eine elektrolytische Behandlung gleich gut geeignet. Welcher von beiden Formen man jeweilig den Vorzug gibt, hängt von den praktischen Verhältnissen ab. Jedenfalls aber werden beide Verfahren häufig und mit Erfolg benutzt.
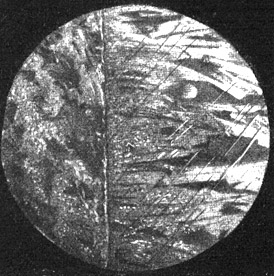
Fig. 107.
Schnitt durch eine Kathode aus reinem Kupferblech (links) mit daraus niedergeschlagenem Elektrolytkupfer (rechts).
Mikrophotographie, hundertfache Vergrößerung. Die Abbildung zeigt (abgesehen von kleinen kristallinischen Verschiedenheiten) links und rechts das gleiche reine Kupfer.
So fanden wir beispielsweise bei unseren Versuchen, eine Kochsalzlösung zu zersetzen, daß es nicht möglich war, die beiden Produkte der Elektrolyse, Natrium und Chlor, zu erhalten, da sie sofort mit dem Wasser der Lösung neue Reaktionen begannen. Hat man es nur auf das Chlor abgesehen, so kann man mit solcher Lösung arbeiten, da schließlich freies Chlor aufgefangen werden kann, nachdem das Wasser einmal mit Chlorgas gesättigt ist.
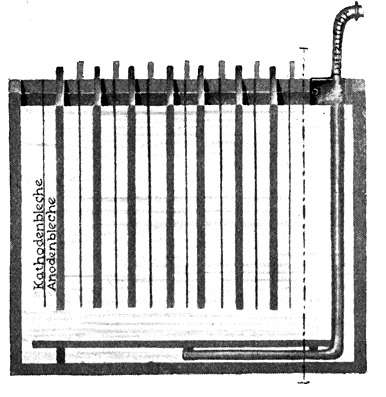
Fig. 108.
Bad zur elektrischen Silberraffinerie (Schnitt), feine Kathodenbleche aus chemisch reinem Silber, dicke Anoden aus gegossenem unreinen Silber.
Die Elektroden stehen in einer Silbernitratlösung. Auch die Silberraffinerie durch Elektrolyse findet heute in ganz großem Maßstabe Anwendung.
Nach diesem Verfahren der Elektrolyse von Kochsalzlösungen wurden denn auch beispielsweise die riesenhaften Chlorgasmengen für den Gaskampf im Weltkriege gewonnen. Will man jedoch das Natriummetall haben, so muß man wohl oder übel zur Schmelze greifen. Man muß das Kochsalz in einem feuerfesten Tiegel bis zum Schmelzpunkt auf 776 Grad Celsius erhitzen. Bei der Elektrolyse scheidet sich dann an der Kathode metallisches Natrium, an der Anode Chlorgas ab. Dies Verfahren krankt aber an dem Mangel, daß metallisches Natrium bereits bei 740 Grad siedet, daß also ein beträchtlicher Teil des gewonnenen Natriums aus dem Tiegel herausdampft, wenn man es nicht durch geschickt angebrachte Vorlagen auffängt. In der Praxis geht man bei der Natriumgewinnung deshalb von der Schmelze des Natriumhydroxyds von der Form NaOH aus, dessen Schmelzpunkt unter dem Siedepunkt des Natriummetalles liegt. Bei der Elektrolyse scheidet sich an der Kathode Natriummetall aus, während an der Anode Oxylgruppen von der Form OH frei werden, die sich zu Wasser und Sauerstoff O2 verbinden. Der Sauerstoff entweicht, während das in die Schmelze gehende Wasser seiner Menge nach gerade ausreicht, um die Hälfte des durch den bisherigen Prozeß gewonnenen Natriummetalls wieder in Ätznatron zurückzuverwandeln. Dies Darstellungsverfahren kann daher höchstens mit einem Wirkungsgrad von 50 Prozent arbeiten, aber es ist die einzige überhaupt in großem Maßstabe brauchbare Methode und wird deshalb in Verbindung mit billigen Wasserkräften zur Natriumgewinnung benutzt.
Während der Bedarf des Weltmarktes an metallischem Natrium verhältnismäßig gering ist, wird das bereits erwähnte Natriumhydroxid oder Ätznatron für viele Fabrikationen, beispielsweise für die Zellulosegewinnung aus Stroh und Holz, in sehr großen Mengen verbraucht. Die Darstellung erfolgt elektrolytisch aus einer konzentrierten Kochsalzlösung, Dabei ist das Bad durch eine poröse Tonwand, ein sogenanntes Diaphragma, in den Kathoden- und Anodenraum getrennt. Die Kathode, an der sich das Ätznatron bildet, besteht aus Eisen, die Anode aus Eisenoxydoxydul, welches vom Chlor nicht angegriffen wird. Bei dieser Anordnung erhält man die Atznatronlösung frei von Chlorgas in dem Kathodenraum.

Fig. 109.
Silberanoden bei der Einhängung in das Bad (starke Platten aus Gußsilber).
Trotzdem ist auch dies Verfahren noch keineswegs ideal schön. Sobald sich nämlich die Kochsalzlauge in dem Kathodenraum genügend mit Ätznatron NaOH angereichert hat, nimmt auch dies an der Elektrolyse teil. An der Kathode sondert sich Natriummetall ab, welches sofort Wasser H2O aufspaltet und wieder Ätznatron NaOH unter Abstoßung von freiem Wasserstoff bildet, hingegen wandern die bei der Elektrolyse des Atznatrons frei werdenden Hyroxylionen durch das Diaphragma zur Anode und bilden dort Wasser und freien Sauerstoff. Man bekommt also infolge dieser sekundären Erscheinungen neben der beabsichtigten Chlor-Natriumzerlegung eine Wasserzersetzung, die natürlich beträchtliche Energiemengen verbraucht und mit wachsendem Ätznatrongehalt immer stärker wird. In der Praxis bricht man die Elektrolyse daher ab, sobald das Kathodenbad etwa 8 Prozent Ätznatron enthält. Diese Ätznatron- und Kochsalzlauge wird dann eingedampft, wobei das Kochsalz bis auf verschwindende Mengen ausfällt und reine Ätznatronlauge abgezapft werden kann. Der Wasserstoff tritt bei diesem Verfahren als ein energieverzehrendes und unerwünschtes Nebenprodukt auf, dessen Menge man durch frühzeitige Unterbrechung des Prozesses möglichst gering zu halten versucht. Immerhin ist die Wasserstofferzeugung beispielsweise in den Bitterfelder chemischen Werken so bedeutend, daß dort alljährlich viele Tausende von Kubikmeter Wasserstoff für die Füllung von Freiballons verwandt werden.

Fig. 110.
Die Silberanoden am Ende des Prozesses.
Die Silberblöcke sind bis auf schwache Reste verschwunden, im Silbernitrat des Bades aufgelöst und an der Kathode wieder niedergeschlagen.
Die Elektrolyse der Alkalien war eine der ersten Taten der jungen Elektrochemie im Anfange des 19. Jahrhunderts. Der englische Physiker Davy, der Entdecker des nach ihm benannten elektrischen Lichtbogens, war im Besitze einer sehr starken Voltaschen Säule und benutzte sie, um die damals noch als Elemente betrachteten Erden Kalk, Kali und Natron in ihrer Schmelze zu elektrolysieren. So entdeckte er in den Jahren von 1803–06 die neuen Metalle Kalium, Natrium und Kalzium. Der elektrische Strom erwies sich schon damals bei seinem ersten Auftreten in der Chemie als ein chemisches Mittel von einer ganz ungeheuren Wirksamkeit. Er war stärker als alle bisher bekannten Reagenzien und Trennungsmittel.
In unserem Zeitalter der billigen Wasserkräfte wird denn auch vom elektronischen Prozeß sowohl in flüssigen Bädern als auch in feurigen Schmelzen in größtem Maßstabe Gebrauch gemacht. Wenn wir heute eine Aluminiumindustrie besitzen, die alljährlich Millionen Kilogramm metallischen Aluminiums gewinnt und zu den verschiedensten Gegenständen, vom Kochtopf bis zum Zeppelingerippe, verarbeitet, so verdanken wir dies der Elektrochemie. Man geht bei der Alumimumgewinnung vornehmlich von einem Mineral, dem Bauxit von der Zusammensetzung Al2O3·2H2O, aus. Dies Mineral ist also ein Hydrat der Tonerde Al2O3. Es ist jedoch so schwer schmelzbar, daß man es mit einem Flußmittel versetzen muß. Hierzu wählt man ein anderes Mineral, den Kryolith von der Zusammensetzung 3NaF·AlF3. Der Kryolith ist also ein Natrium-Aluminiumfluorid. Es spielt bei dem elektronischen Prozeß, wie bereits gesagt, nur die Rolle eines Flußmittels, in welchem sich der Bauxit auflöst. Durch den elektrischen Strom wird zwar das Natriumfluorid zum Teil ebenfalls zersetzt, aber bei der hohen Temperatur der Schmelze vereinigen sich die Komponenten sofort wieder zu Natriumfluorid, und an den Kohlenelektroden des Aluminiumofens scheidet sich einerseits metallisches Aluminium aus, andererseits Sauerstoff, welcher die Kohlenanode allmählich verzehrt.
Das Aluminium ist besonders durch sein geringes spezifisches Gewicht von 2,58 ausgezeichnet. Wöhler stellte es 1827 auf chemischem Wege her, indem er Aluminiumchlorid zusammen mit elektrolytisch gewonnenem Natriummetall erhitzte. Es trat dabei eine Umsetzung in metallisches Aluminium und Kochsalz ein. Das nach diesem Verfahren gewonnene Aluminium stellte sich für das Kilogramm aber auf 2500 Mark, war ziemlich genau so teuer wie Gold. Heute steht der Marktpreis des Aluminiums dagegen auf etwa einer Mark pro Kilogramm. In der Mitte des vorigen Jahrhunderts mußte Napoleon III. seinen Plan, die französischen Kürassiere mit Aluminiumpanzern auszurüsten, aufgeben, weil das Material unerschwinglich teuer war. Heute verdrängt das Aluminiumkabel sogar das Kupferkabel der elektrischen Freileitungen, weil es häufig billiger als dieses wird.
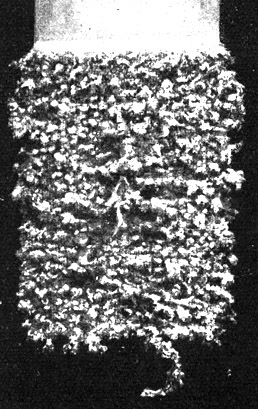
Fig. 111.
Der Niederschlag des elektrolytischen Silbers auf dem feinen Kathodenblech.
Das Silber hat die Neigung, baumartig (Dendriten) herauszuwachsen. Die Kathoden müssen daher öfter aus dem Bade genommen und glatt gewalzt werden.
Zu den elektrolytisch gewonnenen Leichtmetallen gehört auch das Magnesium mit dem spezifischen Gewichte 1,75. Es wird aus dem Karnallit, einem Staßfurter Abraumsalz von der Form MgCl2·KCl, gewonnen. Hier wird also die Verbindung von Magnesiumchlorid und Kaliumchlorid als feurige Schmelze im eisernen Tiegel, der gleichzeitig als Kathode dient, elektrolytisch zerlegt. Das Magnesium wird einerseits als photographisches Blitzlicht viel verwandt. Andererseits bildet es mit dem Aluminium eine wertvolle, als Magnalium bekannte Legierung, welche in der Technik weitgehendste Verwendung findet.
Soweit sich die elektrolytischen Prozesse in Lösungen vollziehen, darf man sicher sein, es mit elektrochemischen Vorgängen zu tun zu haben. Soweit sie sich jedoch in feurigen Schmelzen, also im elektrischen Ofen vollziehen, ist dies nicht ohne weiteres sicher. Die Gewinnung des Aluminiums und anderer Metalle aus den Schmelzen ihrer Verbindungen ist selbstverständlich ein elektrochemischer Vorgang. In vielen anderen Fällen aber, beispielsweise bei der Zusammenschmelzung von Kalk und Kohle zu Kalziumkarbid oder von Silizium und Kohle zu Karborund, handelt es sich um reine Wärmewirkungen, bei denen der elektrische Strom lediglich als Heizmittel dient. Man kann für diese Prozesse daher auch ohne weiteres Wechselstrom benutzen, was für wirkliche Elektrolysen ganz unmöglich wäre.

Fig. 112.
Der Kalziumkarbidofen.
Kein elektrolytischer, sondern ein elektrothermischer Prozeß. Nur die Hitze von 3000°C löst die chemischen Reaktionen aus. Der kohlensaure Kalk wird gebrannt und zu Kalziumoxyd, das mit dem glühenden Kohlenstoff (Koks) derart in Wechselwirkung tritt, daß Kalziumkarbid C2Ca und Kohlensäure CO2 entstehen.
Das gleiche gilt von den elektrischen Stahlöfen, in denen hochwertiger Edelstahl erzeugt wird. Man bedient sich hier des elektrischen Stroms, weil er frei von allen Verunreinigungen dem zu verarbeitenden Material nur die gewünschte Wärmeenergie zuführt. Aber auch nach Ausscheidung dieser elektrothermischen Verfahren bleiben noch überaus zahlreiche elektrochemische Methoden übrig, nach denen die chemische Industrie unserer Tage mit einem Aufwand von Milliarden von Kilowattstunden arbeitet.